

빠르고 정확한 유전체 희귀변이 연관성 분석 방법 개발
- SAIGE-GENE+ improves the efficiency and accuracy of set-based rare variant association tests -
[연구필요성]
바이오 데이터와 임상정보가 결합된 바이오뱅크는 비약적으로 발전하고 있다. 특히 새로 구축되고 있는 대규모 유전체 시퀀싱 데이터는 질병과 연관된 희귀유전변이 발견을 가능케 함으로써, 질병 위험도 예측 및 치료법 발견에 기여할 것으로 기대된다. 하지만 이러한 대규모 데이터의 분석방법, 특히 희귀유전변이와 질병과의 관계를 테스트하는 방법은 잘 정립되어 있지 않다. 현재 존재하는 방법은 몇십만 명의 대규모 데이터를 분석하기에는 너무 느리고, 또 부정확한 추론을 하는 문제가 있다. 일 예로 하나의 유전자인 TTN을 테스트하는데 164 CPU 시간과 65G 바이트 메모리가 필요했으며, 이러한 많은 계산량은 다양한 조건에서 연관성 분석을 하는 것을 막는 요인이 된다.
[연구성과/기대효과]
바이오뱅크의 대규모 시퀀싱 데이터에서 희귀유전자 분석을 위해 우리 그룹은 새로운 SAIGE-GENE+ 방법을 개발하였다. 이전의 방법과 비교해서 SAIGE-GENE+는 계산 시간과 메모리 사용량이 최대 1400배 및 30배 향상되었으며, 가설검정도 정확하게 수행한다. SAIGE-GENE+을 영국의 대규모 바이오뱅크인 UK-Biobank의 20만명 전장 엑솜 데이터에 적용해서 170여 개의 표현형과 18,372개의 유전자를 분석하였고, 이를 통해서 551개의 유전자 표현형 연관성을 발견하였다. 그리고 이러한 분석을 통해 유전변이를 빈도와 기능에 따라서 여러 개의 그룹으로 나눠서 분석한 뒤 합치는 방식이 검정력이 높다는 사실도 발견하였다. 본 연구는 대규모 시퀀싱 데이터에서 희귀유전자 분석이 가능한 새로운 방법을 개발하였고, 이는 희귀유전자와 질병의 연관성을 발견하는 데 공헌하게 될 것이다.
[본문]
인간의 염기서열을 동정하는 차세대시퀀싱방법 (Next Generation Sequencing, NGS)은 급속히 발전하고 있으며, 이에 따라서 대규모 시퀀싱 스터디가 진행되고 있다. 이러한 데이터는 전자의무기록 등의 임상정보 및 다른 바이오 데이터와 결합된 바이오뱅크로 발전하고 있고, 이는 정밀의학을 위한 핵심적인 자원으로 간주되고 있다. 일례로 영국의 바이오뱅크인 UK-Biobank는 47만 명의 대규모 전장 엑솜 유전체 (whole-exome sequencing, WES) 데이터를 발표 하였고, 임상, 이미지, 다른 오믹스 데이터와 결합함으로써 질병의 유전적 기반 발견뿐만 아니라, 질병의 위험도 예측 및 새로운 약물 후보군 발견 등으로도 이어질 수 있다.
DNA 칩에 비교해서 시퀀싱 데이터는 모든 유전변이, 특히 빈도가 1% 이하인 희귀유전변이를 찾고 동정할 수 있다는 중요한 장점이 있다. 이는 인간 유전체에 있는 변이의 대부분 (99% 이상) 이 희귀유전변이 이고, 단백질의 기능을 바꾸는 기능성 유전변이가 자연선택에 의해서 대부분이 희귀유전변이라는 점에서 매우 중요하다. 그러나 몇십만 명의 대규모 시퀀싱 데이터는 이제 만들어지기 시작했으며, 아직은 빅데이터에서 희귀변이를 테스트 하는 방법은 잘 정립되어 있지 않다. 이전에 우리 연구그룹은 희귀유전변이 분석을 위한 SAIGE-GENE 방법을 개발하였다. SAIGE-GENE은 불균형한 질병-대조군 비율의 표현형에 대한 유전자 단위의 희귀유전변이 검사를 수행할 수 있는 유일한 방법이지만, 이 방법도 대규모 UK-Biobank 데이터에 적용되었을 때 많은 문제점을 도출하였다. 연구자들이 매우 희귀한 유전변이(ultra-rare variant)로 테스트를 제한할 경우에 거짓양성 비율이 증가했으며, 계산 비용도 높아서, 예를 들어, 16,227개의 변이가 있는 가장 큰 유전자인 TTN을 테스트하려면 164 CPU hour와 65Gbyte 메모리가 필요했다.
바이오뱅크의 대규모 시퀀싱 데이터에서 희귀유전변이 분석을 위해 우리 그룹은 새로운 SAIGE-GENE+를 개발하였다. 이 새로운 도구는 아주 희귀한 유전변이를 합치는 collapsing 방식으로 변수를 축소하였고, 이는 더 정확한 가설검정을 가능하게 해 주었다. 또한 데이터의 sparsity의 활용 및 C++를 이용한 코드 최적화를 통해서 계산 시간을 크게 단축하였다. 일 예로 TTN을 분석하기 위한 계산 시간과 메모리 사용량은 SAIGE-GENE에 비해 각각 1400배 및 30배(0.11 CPU 시간 2.1G 바이트) 감소했다. 이러한 계산량의 감소는 다양한 조건 (유전변이 빈도수, 유전변이 기능성)에서의 테스트 수행을 가능하게 해 주었다.
SAIGE-GENE+는 UK-Biobank의 전장 엑솜 데이터에 적용되어서 170여개의 표현형과 18,372개의 유전자를 분석하였고, 이를 통해서 551개의 유전자 표현형 연관성을 발견하였다. 우리는 저혈량증과 NOC2L 및 폐경기 연령과 CHEK2의 연관성을 포함한 여러 새로운 유전자와 표현형의 연관성을 확인하였다. 그리고 이러한 분석을 통해 유전변이를 빈도와 기능에 따라서 여러 개의 그룹으로 나눠서 분석한 뒤 합치는 방식이 검정력이 높다는 사실도 발견하였다.
요약하면, 본 연구는 바이오뱅크의 대규모 시퀀싱 데이터에서 희귀유전자 분석을 가능케 하는 새로운 방법을 개발하였고, 이는 희귀유전자와 질병의 연관성을 발견하는 데 공헌하게 될 것이다.
본 연구의 공동 제1저자인 Dr. Wei Zhou, Dr. Wenjian Bi 와 Dr. Zhangchen Zhao 는 연구 책임자인 이승근 교수의 이전 박사과정 학생 (Dr. Wei Zhou, Dr. Zhangchen Zhao) 및 박사 후 연구원 (Dr. Wenjian Bi)이며 현재 Dr. Wenjian Bi 는 중국의 베이징대학교 교수로 재직 중이다.
본 연구는 현재 유전체 연구 분야의 최상위 저널인 Nature Genetics에 개재 승인 받았다.
[연구결과]
SAIGE-GENE+ improves the efficiency and accuracy of set-based rare variant association tests
Wei Zhou*, Wenjian Bi*, Zhangchen Zhao*, Kushal K. Dey, Karthik A. Jagadeesh, Konrad J. Karczewski, Mark J. Daly, Benjamin M. Neale, and Seunggeun Lee
(Nature Genetics, in press)
Several biobanks, including UK Biobank (UKBB), are generating large-scale sequencing data. An existing method, SAIGE-GENE, performs well when testing variants with minor allele frequency (MAF) ≤ 1%, but inflation is observed in variance-component set-based tests when restricting to variants with MAF ≤ 0.1% or 0.01%. Here we propose SAIGE-GENE+ with greatly improved type I error control and computational efficiency to facilitate rare variant tests in large-scale data. We further show that incorporating multiple MAF cutoffs and functional annotations can improve power and thus uncover novel gene-phenotype associations. In the analysis of UKBB WES data for 30 quantitative and 141 binary traits, SAIGE-GENE+ identified 551 gene-phenotype associations.
[용어설명]
- ○유전체중 단백질 정보를 가지는 엑손을 모두 포괄한 영역
[그림설명]
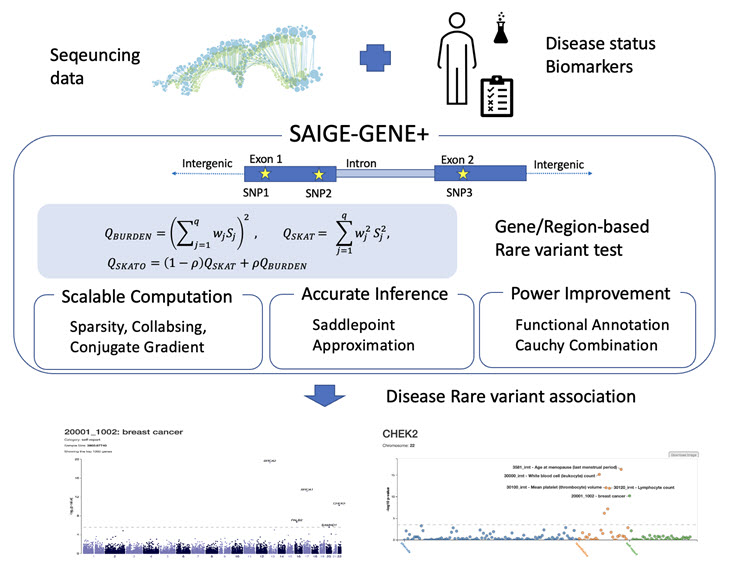
SAIGE-GENE+ Overview